References:
Huang, Y.-T., Chao, K.-M.,
and Chen, T., 2005, “An
Approximation Algorithm for Haplotype Inference by Maximum Parsimony,”
Journal of Computational Biology,
12: 1261-1274.
PowerPoint Presentation
Given a set of genotypes, we wish to find a minimum subset of haplotypes that can resolve these genotypes. This problem has been shown as NP-hard.
|
|
For short genotypes, we design and implement a software called SDPHapInfer. |
|
|
For long genotypes, we design and implement a software called LongHapInfer. |
Note that the zip files are compressed using WinZip. If you encounter any problem, please contact Prof. Kun-Mao Chao.
SDPHapInfer is implemented in MatLab. The SDPHapInfer formulates the haplotype inference problem as the following integer quadratic programming (IQP) problem. The objective function is to minimize the number of haplotypes, and the constraint functions are to resolve each genotype.
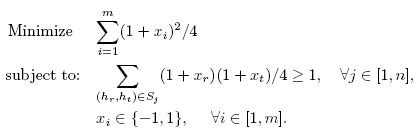
To solve the IQP problem, we propose an iterative SDP-relaxation algorithm which finds a solution within a factor of O(log n) of the optimal solution. The SDP problem is solved via an external MatLab library called SDPA-M.
|
|
Download SDPHapInfer. This zip file contains four MatLab scripts: SDPHapInfer.m, ReadGenoFile.m, Invert.m, Chk_Resolve.m, and Find_Index.m. |
|
|
Input file format: |
|
|
To run this program, type
"SDPHapInfer" under the MatLab command line, and you will see a prompt for
input of the file name. |
|
|
The SDPHapInfer will generate an
output file *.out (e.g., test.dat.out) that contains solutions. |
LongHapInfer is implemented in MatLab. This algorithm partitions the genotypes into segments, infers subhaplotypes for each segment, and concatenates subhaplotypes in different segments for the final solution. LongHapInfer requires an external MatLab library called SDPA-M.
|
|
Download LongHapInfer. This zip file contains four MatLab scripts: LongHapInfer.m, ReadGenoFile.m, Invert.m, Chk_Resolve.m, and Find_Index.m. |
|
|
Input file format: |
|
|
To run this program, type
"LongHapInfer" under the MatLab command line, and you will see a prompt
for
input of the file name. |
|
|
The LongHapInfer will generate an output file *.out (e.g., test.dat.out) that contains solutions. |
